3.1 FlyBase y GBrowse
Los elementos transponibles o elementos móviles del genoma son un tipo de mutación que consiste en secuencias de ADN que saltan de un sitio a otro del genoma. A cada elemento transponible, cuando se descubre, se le pone un nombre.
En Drosophila melanogaster, todos los TEs se recogen, junto con mucha otra información genética, en una gran base de datos. Esta base de datos se llama FlyBase. Qué original, ¿verdad? Para hacerlo más sencillo a cada TE se lo asigna un ID (identificador), un código único dentro de la base de datos.
La nomenclatura de los TEs en FlyBase es la siguiente: todos los TEs empiezan por FBti (FlyBase Tranposable Insertion) seguidos de un número único. Por ejemplo: FBti0019099.
El GBrowse es un visualizador de información genética de una especie en concreto. Podemos navegar, movernos por los genomas y ver sus características. ¿Lo queréis ver? ¿Por qué no buscamos el ejemplo anterior en el GBrowse de Drosophila? Veamos dónde se encuentra este TE en el genoma y qué genes tiene cerca.
Para ello, hay que ir a la web de FlyBase En el panel superior hay que seleccionar el segundo cuadrado en el que pone GBrowse.
Una vez dentro, en landmark o región podemos escribir el código de nuestro TE de interés, ej. FBti0019099.
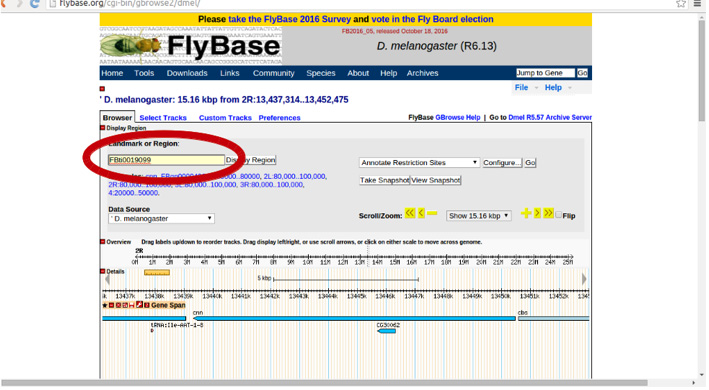
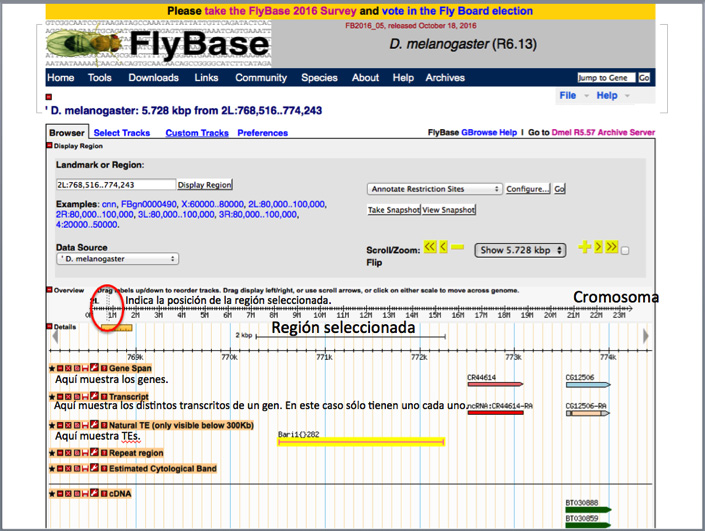
Mediante la pestaña del zoom podéis ampliar o reducir la región para ver que hay más allá de las ~ 6000 pb que el GBrowse muestra por defecto.
Observación – Medidas Genómicas
- Un par de bases (bp, en inglés) es un par de nucleótidos «enfrentados», e indica una posición única en el genoma.
- Una kilobase (kb) son 1.000 bp.
- Una megabase (Mb) son 1.000 kb o 1.000.000 bp.
Podéis explorar y jugar con el navegador. Si en algún momento no sabéis cómo volver atrás, volver a poner el ID del TE (FBtiXXXXXXX) del TE en la casilla de búsqueda y volveréis a los ~ 6.000 pb iniciales.
3.2 Archivo de datos, ¿qué contiene?
En #MelanogasterCTF ponemos a vuestra disposición un archivo de datos que contiene la lista de un grupo de TEs del genoma de Drosophila melanogaster. Como hemos dicho, la mayoría de las mutaciones que se producen son deletéreas y, por lo tanto, la mayoría estarán a frecuencias muy bajas. Es decir, pocos individuos (a veces solo uno o dos) tendrán esas mutaciones. Se estima que hay unos ~ 20.000 TEs en cada una de las poblaciones de Drosophila. Las herramientas científicas actuales nos permiten detectar y estimar la frecuencia en la población de forma fiable en un total de 1632 TEs, ya que estos son secuencias altamente repetitivas y por tanto son difíciles de distinguir unos de otros. Sin embargo, debemos recordar que 1632 no son la totalidad de TEs existentes, sino la cantidad con la que podemos trabajar de forma fiable hoy en día.
La generación de científicos anterior a la nuestra solo pudo analizar casos concretos. Ahora podemos analizar 1632 TEs de forma conjunta. Quizá, cuando vosotros seáis científicas y científicos ¡podréis analizar los ~20.000 TEs!
El archivo de datos contiene por lo tanto 1632 TEs y la siguiente información en cada columna:
- ID del TE (TE_ID)
- su nombre (Tename)
- el cromosoma (chr) en que se encuentran en el genoma.
- la posición inicial en la que se encuentran en el genoma (start).
- la posición final en la que se encuentran en el genoma (end).
- los niveles de recombinación en la región del genoma en la que se encuentran.
- distancia en bp al gen más cercano. Cuando la distancia es 0 significa que el TE está dentro o solapando con el gen más cercano. Podéis probad los ejemplos siguientes buscando en el GBrowse: FBti0019985 o FBti0019017.
- el ID de uno de los genes más cercanos
- el nombre de ese gen
- – 14. las frecuencias a las que se encuentran los TEs en 5 poblaciones. Una población africana, de Zambia (columna 10), de cuyos ancestros se considera que provienen las moscas europeas y 4 poblaciones europeas: Portugal (Recarei) (columna 11), España (Gimenells) (columna 12), Alemania (Múnich) (columna 13) y Finlandia (Akaa) (columna 14).
Guárdalo localmente en tu ordenador, ¡y vamos a trabajar con él!
3.3. ¿Cómo importar el archivo de datos a R?
Para empezar a trabajar hay que introducir el archivo de datos en R.
Para ello podéis usar la función que se encuentra en el archivo de código R. La función que os permitirá importar el archivo de datos es la siguiente:
Nombre_archivo <- read.delim (file=”path_archivo”, head=TRUE)
En el archivo de código que os habéis bajado anteriormente (módulo 2.4) podéis encontrar los ejemplos reales de como usar las funciones. Tendréis que copiar la función a vuestro archivo de código propio en el editor de texto y cambiar el nombre del archivo y la ruta (el path) a donde os habéis descargado o guardado el archivo de datos. Luego podréis copiarlo y pegarlo en la “consola” de R (recuadro izquierdo rojo).
Las condiciones en R se expresan de la siguiente manera:
- Igual que es ==
- Distinto de es ¡=
- Mayor que es >
- Menor que es <
- Para realizar múltiples condiciones se utiliza & (y) o | (o).
Si con la función no lo conseguís también podéis importar el archivo de la siguiente manera:
Usar la pestaña de Import Dataset en el recuadro superior derecho “Environment”. Seleccionar la opción de CVS.

Pulsar sobre Browse y seleccionar el archivo de datos.
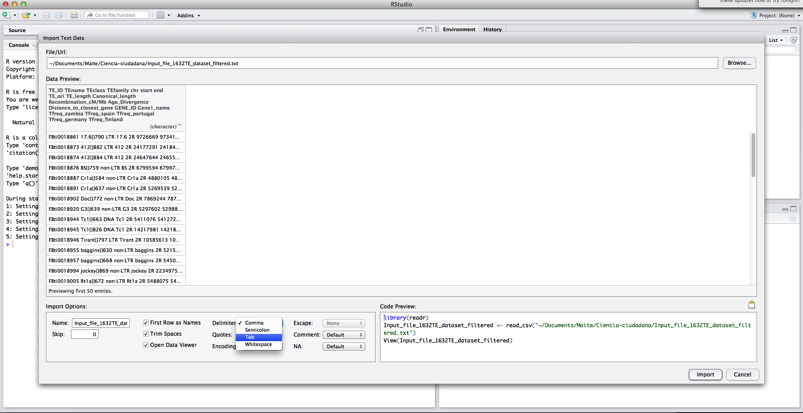
Por último, tendréis que cambiar el delimitador a tabulador (tab) y cambiar la opción de “Quotes” a “none”.

Pulsar sobre Import
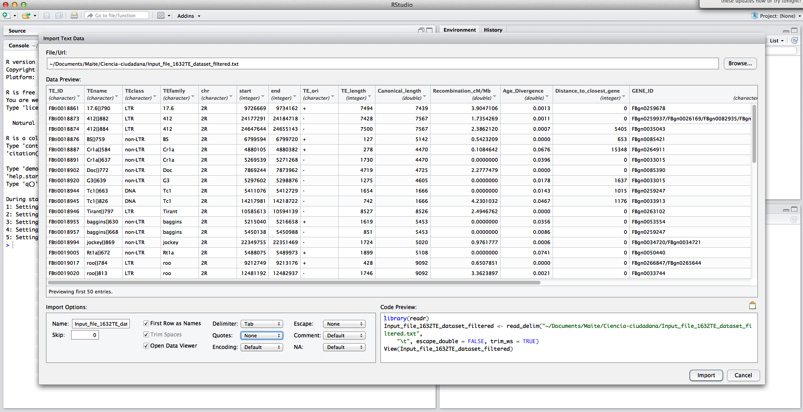
Si lo habéis hecho correctamente veréis vuestro archivo en la ventana superior izquierda del programa. ¡¡Y ya podéis empezar a trabajar en R!!
Aquí tenéis una breve descripción de la función de cada pantalla y la información proporcionada.
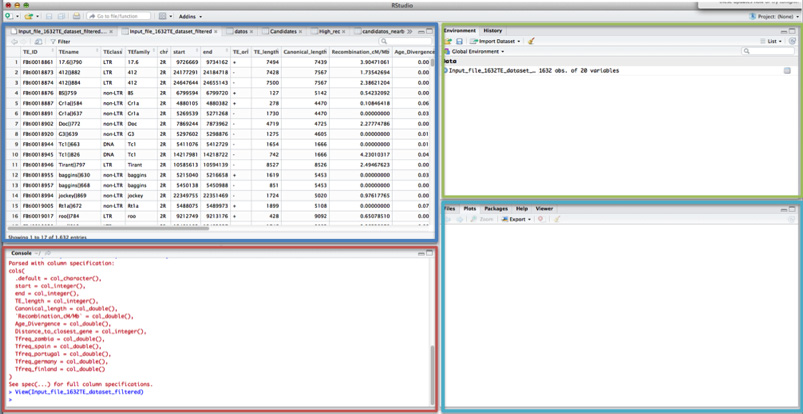
La pantalla más importante es el RECUADRO ROJO: la consola o terminal. Allí copiaréis y pegaréis todos los comandos que queráis ejecutar en R.
RECORDAD: Las condiciones en R se expresan de la siguiente manera:
- Igual que es ==
- Distinto de es ¡=
- Mayor que es >
- Menor que es <
- Para realizar múltiples condiciones se utiliza & (y) o | (o).
Aunque RStudio permite guardar los comandos ejecutados desde esta terminal, os aconsejamos que copiéis los comandos definitivos en un archivo aparte, abierto y guardado con un editor de texto. Así, podréis seguir fácilmente trabajando si lo hacéis en días o ordenadores diferentes. Tendríais que tener vuestro propio archivo de código, como el que os proporcionamos, pero personalizado. Así, simplemente tendréis que copiar y pegar todos los comandos y podréis seguir trabajando en el punto que estabais.
En el RECUADRO AZUL podéis visualizar la información del archivo de datos. En distintas pestañas irán apareciendo los nuevos datasets que introduzcáis o creéis.
En el RECUADRO VERDE tenéis 2 pestañas:
- Environment, donde entre otras cosas podréis ver los distintos datasets que abráis en R o vayáis generando.
- History, donde se guardan todos los comandos que uséis, los de prueba, los que funcionan y los que no funcionan.
Por eso os recomendamos que una vez funcione un comando lo guardéis en un archivo de texto aparte, y así generéis vosotros mismos vuestro Historial con solo los comandos que funcionen realmente o sean importantes para el trabajo.
En el RECUADRO TURQUESA, hay varias pestañas. Si seleccionáis la pestaña “Plots” podréis ver aquí mismo las figuras que vayáis generando.
3.4. Distribución de las variables de interés
Ahora que los datos están correctamente introducidos podremos analizar la distribución de las variables que tenemos en el archivo de datos.
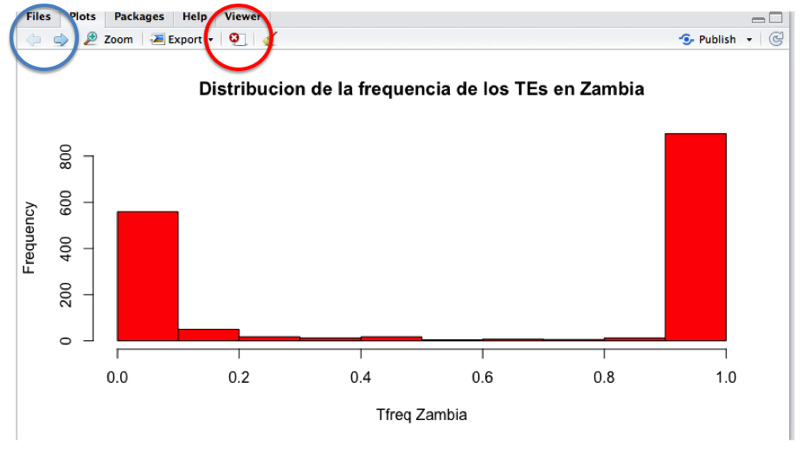
¿Sabéis cual es la distribución de la frecuencia de los TEs en las 5 poblaciones secuenciadas? Hagamos un histograma para verlo más fácil.
hist (Nombre_archivo$Tfreq_zambia)
Encontrarás cómo se usa esta función en el ejemplo real en el archivo de código en el editor de texto. Tienes que sustituir el nombre que le hayas puesto al archivo de datos.
¡Ya lo tenemos! ¡Pongámoslo un poco más bonito, démosle color!
hist (Nombre_archivo$Tfreq_zambia, main=”el título del gráfico”, xlab=”nombre_variable”, col=”red”)
Encontrarás como se usa esta función en el ejemplo real en el archivo de código en el editor de texto.
Puedes elegir los colores en este enlace.
Si quieres seguir probando y haciendo el gráfico más y más completo puedes echar un vistazo a esta otra página.
Ahora veamos cómo es la distribución de los TEs en otras poblaciones. Cada gráfico aparece en el mismo recuadro y con las flechas en la parte superior del recuadro podrás desplazarte de uno a otro. Con el símbolo de la x (“remove the current plot”) podrás borrar los gráficos.